

Inside each one of your cells, there’s enough DNA to stretch more than two metres if laid out end-to-end. Yet somehow, all this DNA fits into a tiny structure called the cell nucleus, so intricately folded and tightly packed that it makes origami seem simple by comparison. But scientists are learning that this incredible folding isn’t just a clever trick to save space; it may also help cells make critical decisions about what kind of cells they become and how they behave.
A study published in the Bulletin of Mathematical Biology by researchers Daria Stepanova (CRM), Meritxell Brunet Guasch (University of Edinburgh), Helen M. Byrne (University of Oxford), and Tomás Alarcón (ICREA–CRM) explores how DNA’s 3D folding influences gene activity. They discovered that the shape of DNA, and how molecules interact with it, may be key to determining whether genes are switched “on” or “off.”
This switching of genes on and off without changing the DNA code is known as epigenetics. Epigenetic changes occur through chemical marks that attach to the DNA molecule or to proteins called histones, around which DNA wraps. Think of these marks as sticky notes placed along the DNA: they don’t change the instructions in our genetic code, but they tell cells which parts of those instructions to read or ignore. These notes are crucial for guiding a cell’s identity, allowing it to specialise into muscle, nerve, skin, or blood cells, even though every cell begins life with the same DNA.
However, a key mystery remains: sometimes these epigenetic marks form smooth patterns across the genome, while other times they become strikingly uneven or “rugged,” marked by abrupt shifts between areas where genes are active and areas where they’re silenced.
Until now, exactly how and why these complex, rugged patterns emerge has not been well understood.
A Virtual Lab Built with Mathematics
To solve this mystery, the research team created a mathematical model, a kind of “virtual lab,” that allows them to test biological rules and see what patterns emerge through computer simulations. Their model specifically explored the interactions between key histone modifications and the enzymes responsible for adding (“writing”) or removing (“erasing”) these chemical marks.
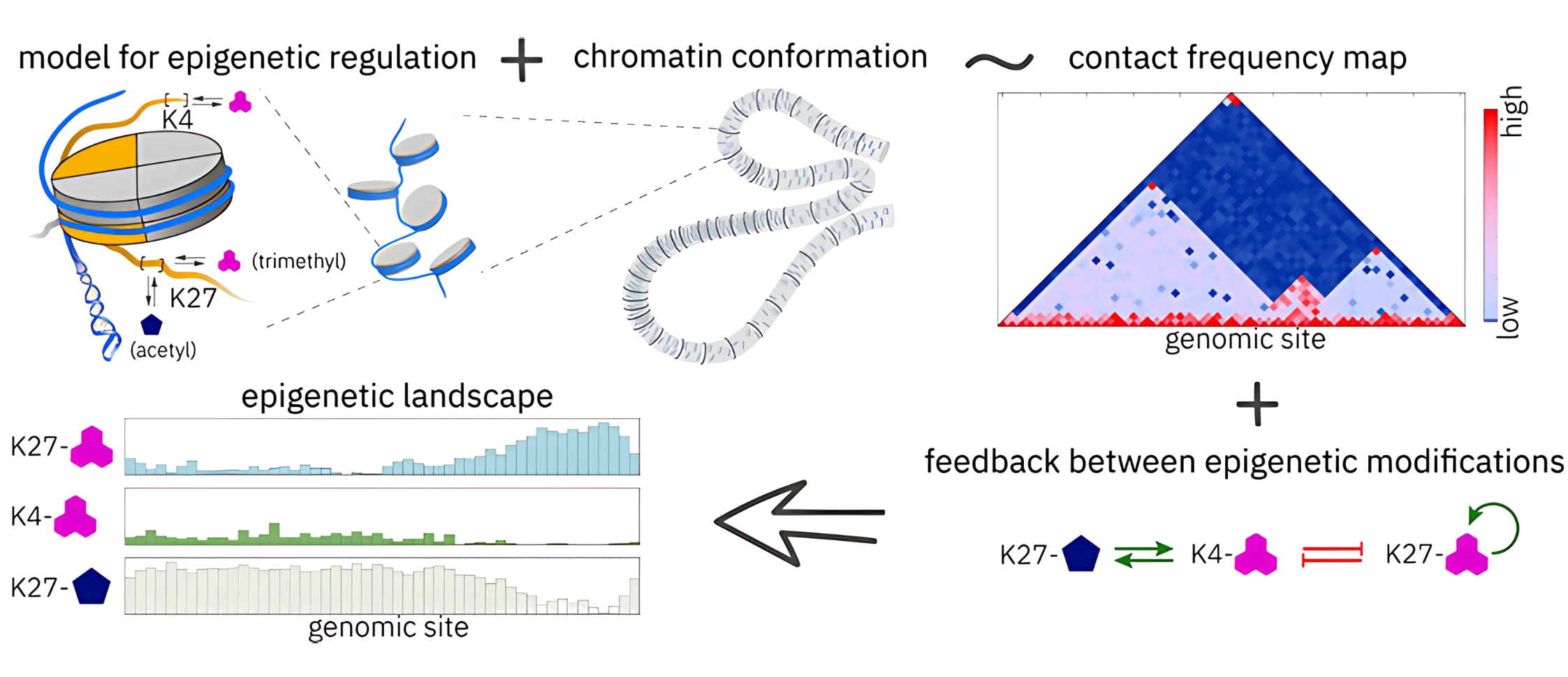
The model considers epigenetic modifications (H3K27me3, H3K4me3, H3K27ac) on histone H3 residues. It divides chromatin into fixed-size genomic regions characterised by these modifications, linked through reinforcing and inhibitory enzyme-driven feedback.
What set their approach apart was the inclusion of two critical elements usually overlooked in other studies. First, they included the actual 3D folding structure of chromatin (DNA wrapped around histones) because how chromatin folds changes which parts of the genome interact. Second, they factored in the limited availability and competition among enzymes. These enzymes constantly “fight” over which sites on the chromatin they modify, creating a microscopic molecular tug-of-war.
“There are not so many models of epigenetic regulation, especially those which also account for chromatin conformation; therefore, previous models have been formulated in a more simplified setting.” As Stepanova explains, “enzyme competition can be important in shaping epigenetic landscapes. It is a more realistic assumption than a constant or uniform concentration of enzyme along the chromatin. Chromatin is folded, and enzyme molecules are ‘floating’ around, ready to bind to any genomic site in their vicinity.” Moreover, she notes, this enzyme competition can “break the symmetry and eventually lead to the emergence of rugged epigenetic patterns.”
By combining chromatin folding and enzyme competition, the team found that remarkably uneven patterns emerged. When distant segments of DNA come into close contact due to folding, they influence each other’s epigenetic states. At the same time, because the enzymes needed to modify these regions are limited, only some areas get marked, leaving others untouched. The result is a patchwork, or rugged landscape, of active and inactive zones that mirrors what scientists see in real cells.
Stepanova further highlights why identifying these rugged and uniform regimes is crucial: “Epigenetic marks are known to have self-reinforcing feedback mechanisms, promoting the addition of the same or similar marks in neighbouring chromatin regions. If the region is extended, these loops would make the entire region uniform. This does not coincide with reality, since we know that, especially in differentiated cells, there are chromatin regions which are silenced and chromatin domains which are active.”
She notes that this raises an important question: “What is the ‘stopping factor’ that maintains the boundaries between these activating and inhibitory regions?” Their research provides “a possible explanation for one of the factors contributing to the emergence of alternating silenced and active genomic regions.”
Interestingly, the team’s model also captured another aspect observed in real cells called bivalent chromatin. These are regions carrying both activating and silencing epigenetic regulators at once, like genes being kept on standby, ready to quickly turn “on” or “off” in response to specific signals. These bivalent states help cells transition smoothly from uniform epigenetic landscapes to more rugged patterns, or vice versa.
Bridging Scales
The researchers designed their model to function effectively across different scales, from individual gene regions to larger chromatin structures known as Topologically Associating Domains (TADs). This mesoscale approach is particularly important. “Higher-order structures, such as loops and small TADs, can be visualised more precisely, and we can investigate how these structures influence the formation of epigenetic patterning,” Stepanova explains. “If we go to large scales of the entire chromosome, these details are also lost. In my view, the mesoscopic approach is ideal for exploring epigenetic landscapes.”
Stepanova also highlights the unique advantages mathematics offers in studying epigenetics. “Using mathematical modelling, we can investigate various aspects of biological processes which cannot be manipulated, visualised, isolated, measured, or controlled in an experimental setting,” she notes. While experimental studies are powerful, they can be costly and time-consuming. Theoretical modelling, she points out, is essentially cost-free, though closer collaboration with biologists is essential.
As an example of this type of collaboration, Stepanova mentions the current work of her master’s student Roger Bosch, who is investigating how various methods of processing chromatin conformation data affect our understanding of epigenetic patterns. These techniques aim to remove experimental artefacts and clarify the true chromatin folding patterns. However, Stepanova remarks, “It is still unclear how much these methods give us a prettier picture and how much they genuinely bring us closer to the accurate structure of cellular DNA.”
Questions like these underline both the importance and potential of integrating theoretical modelling closely with experimental research to improve our understanding of biological data.
Citation:
Stepanova, D., Brunet Guasch, M., Byrne, H.M. et al. Understanding How Chromatin Folding and Enzyme Competition Affect Rugged Epigenetic Landscapes. Bull Math Biol 87, 59 (2025). https://doi.org/10.1007/s11538-025-01434-0
crm researchers

Tomás Alarcón studied Physics, obtaining his PhD in 2000. He then pursued postdoctoral research at Oxford’s Centre for Mathematical Biology, collaborating with Helen Byrne and Philip Maini on mathematical models of tumour growth, a topic he continues researching today. After further postdoctoral positions at University College London and Imperial College, focusing on tumour dormancy, receptor dynamics, and epigenetic evolution, he led the Mathematical Biology group at the Basque Centre for Applied Mathematics (BCAM). Since 2010, he has headed the Computational & Mathematical Biology group at CRM.

Daria Stepanova is a researcher specialising in mathematical modelling applied to theoretical biology. She completed her PhD at the CRM (UAB) under the supervision of Tomás Alarcón (ICREA–CRM), Helen Byrne, and Philip Maini (University of Oxford), focusing on blood vessel growth (angiogenesis). After a research stay at Oxford studying cellular interactions linked to cancer, she joined the Laboratorio Subterráneo de Canfranc (LSC), providing mathematical support for experiments such as Hyper-Kamiokande and investigating how low-background radiation affects biological systems. Currently back at CRM as a postdoc, she continues exploring the complex interactions between epigenetic regulation and the 3D chromatin structure.
Subscribe for more CRM News
|
|
CRM CommPau Varela
|

CRM Awards Its Prize at Exporecerca Jove for the Third Time
For the third year running, CRM visited Exporecerca Jove to award its prize to the student project with the strongest mathematical content. This edition, the jury selected two winners: Xavier Ortiz Quintana, who built a real-time 3D scanner using...

When Symmetry Breaks the Rules: From Askey–Wilson Polynomials to Functions
Researchers Tom Koornwinder (U. Amsterdam) and Marta Mazzocco (ICREA-UPC-CRM) published a paper in Indagationes Mathematicae exploring DAHA symmetries. Their work shows that these symmetries shift Askey–Wilson polynomials into a continuous functional setting,and...

Homotopy Theory Conference Brings Together Diverse Research Perspectives
The Centre de Recerca Matemàtica hosted 75 mathematicians from over 20 countries for the Homotopy Structures in Barcelona conference, held February 9-13, 2026. Fourteen invited speakers presented research spanning rational equivariant cohomology theories, isovariant...

Three ICM speakers headline the first CRM Faculty Colloquium
On 19 February 2026, the Centre de Recerca Matemàtica inaugurated its first CRM Faculty Colloquium, a new quarterly event designed to bring together the mathematical community around the research carried out by scientists affiliated with the Centre. The CRM auditorium...

Trivial matemàtiques 11F-2026

Rescuing Data from the Pandemic: A Method to Correct Healthcare Shocks
When COVID-19 lockdowns disrupted healthcare in 2020, insurance companies discarded their data; claims had dropped 15%, and patterns made no sense. A new paper in Insurance: Mathematics and Economics shows how to rescue that information by...

L’exposició “Figures Visibles” s’inaugura a la FME-UPC
L'exposició "Figures Visibles", produïda pel CRM, s'ha inaugurat avui al vestíbul de la Facultat de Matemàtiques i Estadística (FME) de la UPC coincidint amb el Dia Internacional de la Nena i la Dona en la Ciència. La mostra recull la trajectòria...

Xavier Tolsa rep el Premi Ciutat de Barcelona per un resultat clau en matemàtica fonamental
L’investigador Xavier Tolsa (ICREA–UAB–CRM) ha estat guardonat amb el Premi Ciutat de Barcelona 2025 en la categoria de Ciències Fonamentals i Matemàtiques, un reconeixement que atorga l’Ajuntament de Barcelona i que enguany arriba a la seva 76a edició. L’acte de...

Axel Masó Returns to CRM as a Postdoctoral Researcher
Axel Masó returns to CRM as a postdoctoral researcher after a two-year stint at the Knowledge Transfer Unit. He joins the Mathematical Biology research group and KTU to work on the Neuromunt project, an interdisciplinary initiative that studies...

The 4th Barcelona Weekend on Operator Algebras: Open Problems, New Results, and Community
The 4th Barcelona Weekend on Operator Algebras, held at the CRM on January 30–31, 2026, brought together experts to discuss recent advances and open problems in the field.The event strengthened the exchange of ideas within the community and reinforced the CRM’s role...

From Phase Separation to Chromosome Architecture: Ander Movilla Joins CRM as Beatriu de Pinós Fellow
Ander Movilla has joined CRM as a Beatriu de Pinós postdoctoral fellow. Working with Tomás Alarcón, Movilla will develop mathematical models that capture not just the static architecture of DNA but its dynamic behaviour; how chromosome contacts shift as chemical marks...

Criteris de priorització de les sol·licituds dels ajuts Joan Oró per a la contractació de personal investigador predoctoral en formació (FI) 2026
A continuació podeu consultar la publicació dels criteris de priorització de les sol·licituds dels ajuts Joan Oró per a la contractació de personal investigador predoctoral en formació (FI 2026), dirigits a les universitats públiques i privades del...